
Want to know more about MPS I Hurler, Hurler Scheie and Scheie? Watch our animated guide.
On this page you can find our animated guide, general information about the condition, latest news, updates and stories and a list of relevant resources and events.
Understanding the condition
What is MPS I?
MPS I is one of the mucopolysaccharide storage diseases. MPS I includes Hurler, Hurler-Scheie and Scheie diseases. These diseases differ in severity of symptoms across a spectrum and are named after the doctors that identified them.
Based on the presence of symptoms Hurler disease was first described by Dr Hurler in 1919, later in 1962 Dr Scheie identified a less severe form and referred to is as Scheie disease.
People with MPS I who appear not to fit clearly at either end of the spectrum of Hurler or Scheie are classified with Hurler-Scheie disease.
Frequently asked questions
Mucopolysaccharides are long chains of sugar molecules used in the building of bones, cartilage, skin, tendons and many other tissues in the body. “Muco” refers to the thick jelly-like consistency of the sugar molecules, “poly” means many, and “saccharide” is a general term for the sugar part of the molecule. In the course of normal life there is a continuous recycling process of building new mucopolysaccharides and breaking down old ones. The breakdown and recycling process requires a series of special biochemical tools called enzymes.
People with MPS I are missing or are low in an enzyme called alpha-L-iduronidase, which is essential in breaking down mucopolysaccharides dermatan sulphate and heparan sulphate. When dermatan sulphate and heparan sulphate are not completely broken down they remain stored in the body. The symptoms of MPS I occur when there is a build-up of dermatan sulphate and heparan sulphate in the tissues in the body. Babies may show little sign of the disease but as more and more cells build up with partially broken down mucopolysaccharides, symptoms start to appear.
MPS I is an autosomal recessive disease this means that both parents must carry the same affected gene and each pass this same affected gene to their child.
People probably carry from 5 to 10 genes with mutations in each of their cells. Problems happen when the particular gene is dominant or when a mutation is present in both copies of a recessive gene pair. Genes are the unique set of instructions inside our bodies that make each of us an individual. They are the blueprint for our growth and development, as well as controlling how our bodies function. Genes are carried on structures called chromosomes and it is usual to have 23 pairs. A child will inherit half of the chromosomes from the mother and the other half from the father resulting in 23 pairs. 22 of these pairs look the same in both males and females. Pair 23 are the sex chromosomes, and this is the pair that differ between females and males. The X chromosome is inherited from the mother and the Y chromosome is inherited from the father. More information about inheritance is available here.
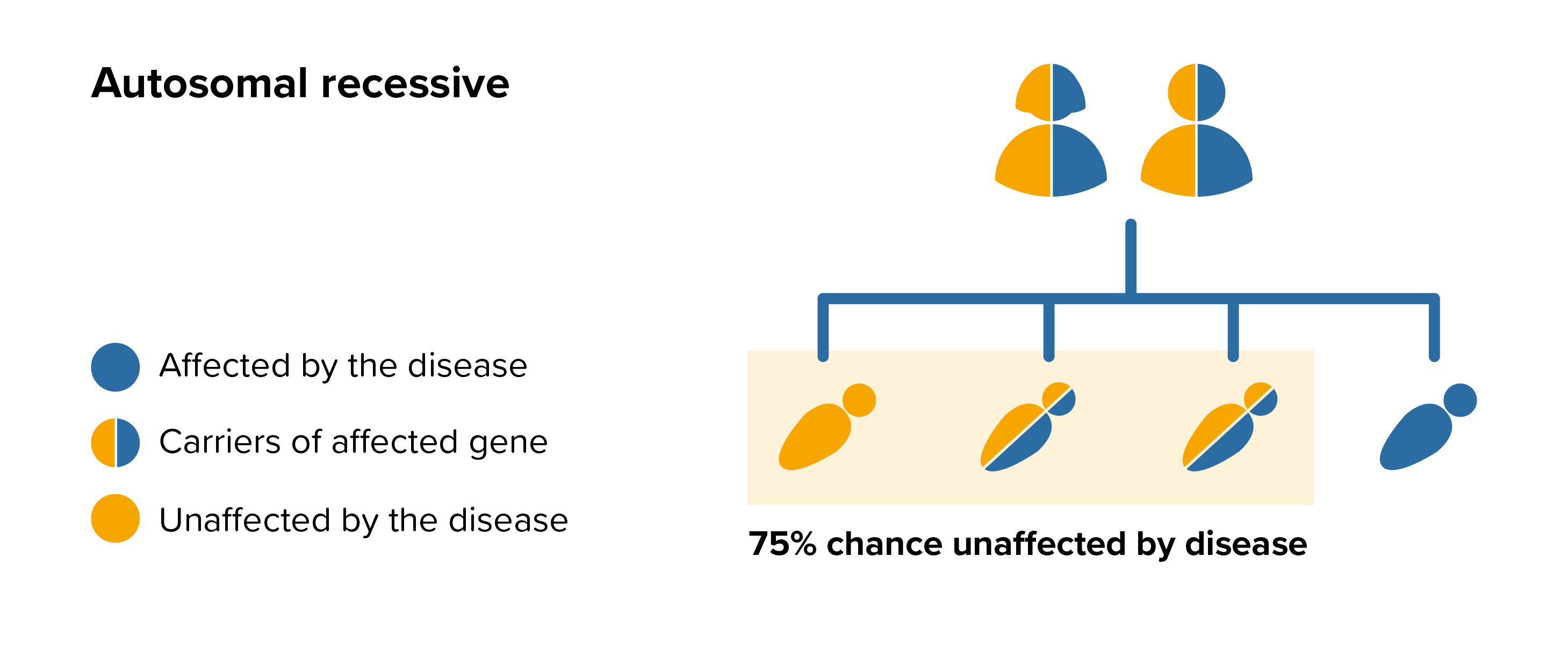 For each pregnancy the chances of a baby inheriting MPS I are completely independent of whether a previous child was affected with MPS I. With each pregnancy there is a 1 in 4 chance that the baby will be affected by MPS I.
For each pregnancy the chances of a baby inheriting MPS I are completely independent of whether a previous child was affected with MPS I. With each pregnancy there is a 1 in 4 chance that the baby will be affected by MPS I.
All parents of children with MPS I can benefit from genetic counselling, the counsellor can provide advice on the risk to close relatives and to suggest whether the wider family should be informed. To find out during a pregnancy, if the baby is affected by MPS I, screening tests can be arranged early on during a pregnancy for those families who already have a child with MPS I. Where only one parent is a carrier, they can opt for carrier screening but it is not 100% reliable or accurate and is not possible in all cases. Amniocentesis and chorionic villus sampling are both available during the pregnancy to find out if the baby is affected by MPS I.
It might also be possible to have Pre-implantation genetic diagnosis (PGD) screening to avoid passing MPS I to the baby. PGD is an assisted fertility treatment that involves checking the chromosomes of embryos before they are transferred in the womb using IVF techniques.
It is estimated that nearly 6% of the UK population (around 3.5million people) will be affected by a rare disease at some point in their lives. A single rare disease may affect up to about 30,000 people however the vast majority of rare diseases affect far fewer than this.
During a 10 year period (1989 to 1999) 88 babies were born with MPS I in the UK.
People with MPS I can experience some or many symptoms from a wide spectrum which range from severe to very mild.
Generally, children with the classic severe form of Hurler disease have progressive developmental delay, severe progressive physical problems and early advancement of the disease.
People with Scheie disease do not have progressive developmental delay and their physical problems advance more slowly. People with Hurler-Scheie disease will fall between the two ends of the spectrum. It is important to note that people with MPS I will not all experience all the symptoms.
For more on the symptoms, see:
Enzyme Replacement Therapy (ERT)
For people with Hurler-Scheie and Scheie diseases ERT is a long-term therapy whereby the missing or deficient enzyme is given via an intravenous infusion. The name for the replacement enzyme in MPS I is laronidase and the brand name is Aldurazyme®. Aldurazyme® was licensed as an ERT in 2003 and has been shown to reduce many of the non-brain related symptoms, such as improving respiratory function and mobility, and reducing joint stiffness.
Aldurazyme® is a weekly infusion lasting 3 to 4 hours that is usually administered at home. More information about Aldurazyme® can be found at www.aldurazyme.com and a UK version of the patient information leaflet is here. Further information on this treatment is available from the electronic medicines compendium.
For people with Hurler disease ERT is administered for a brief time before and after Haematopoietic Stem Cell Transplantation therapy.
Haematopoietic Stem Cell Transplantation (HSCT)
For Hurler disease HSCT is the treatment of choice for children up to 2 years old. The immediate benefits include correction of the missing or deficient enzyme.
The long-term benefits include a longer life by protecting the heart, lungs and brain from the effects of progression of MPS I. Other organs and tissues can also show benefits from the therapy; these include the eyes and ears, liver, spleen, joints and airways. Even after a successful transplant and experiencing several benefits many people with MPS I may still require a range of orthopaedic surgeries.
For an up-to-date list of current UK based trials taking place visit Be Part of Research (resource provided by the National Institute for Health Research). For an international search visit Clinical Trials (resource provided by the U.S. National Library of Medicine).
This resource provides information on trial status including recruiting, completed or withdrawn and worldwide trial locations. To find out more about past or current trials speak to your doctor and learn about the risks and potential benefits.
The MPS Society is the only UK charity at the forefront of supporting people and families affected by MPS and related diseases. Our extensive support services offer you a wide range of support and resources.
The team can advise and sign post you to adequate needs-led support and services in your local area as well as social care, home adaptions, education and much more.
The support team can visit you in your home and provide you with vital support.
Get involved and support us in the community, volunteer or support fundraising; we are a small charity but with your support we can continue to offer a highly valued and essential service.
Latest resources

MPS I Scheie disease - information for individuals, parents and families
We know that being diagnosed with a rare disease is life-changing and you can struggle to come to terms with it. Therefore, we have...

MPS I Hurler-Scheie disease - information for individuals, parents and families
We know that being diagnosed with a rare disease is life-changing and you can struggle to come to terms with it. Therefore, we have...

MPS I Hurler disease - information for individuals, parents and families
We know that being diagnosed with a rare disease is life-changing and you can struggle to come to terms with it. Therefore, we have...
Miya's story
When Miya was 2 weeks old, she began showing signs that worried us. She wouldn’t stop crying and she suffered terribly from “silent reflux”.





Latest news and blogs

News
FDA places temporary clinical hold on two MPS gene therapy trials
The MPS Society is aware of an announcement from REGENXBIO regarding its investigational gene therapy programs RGX-111 for MPS I Hurler syndrome) and RGX-121 for MPS II (Hunter syndrome).
29/01/2026

Condition
The importance of clinical guidelines
Regardless of location and specialist teams, clinical guidelines exist to ensure the same high standard of care. Learn more about our involvement in their development and see which conditions have so far been published.
21/08/2025

Blog
Emma's story | The Big Give Christmas Challenge 2023
Hello, everyone! I am Emma, mum to my wonderful daughter Isabella. Isabella has MPS I Hurler and we have been members of the MPS Society since 2016.
14/11/2023
Your stories

Story
Ayla's story | MPS Awareness Week 2026
This MPS Awareness Week, we hear Ayla’s story. Diagnosed with MPS I Hurler at the age of four, Ayla is at the heart of a close‑knit and outgoing family unit.
13/05/2026

Story
Emily's story | Big Give Christmas Challenge 2025
Emily shares why community events mean so much to our families, from feeling accepted and understood to enjoying a day out away from the everyday challenges and creating new friendships.
04/11/2025

Story
BBC Children in Need’s Greatest Showman
If you followed last year’s BBC Children in Need programme, you simply couldn’t have missed Ethan who joined the One Show’s CIN Challenge Squad and starred as The Greatest Showman in the live show. Hear all about his incredible experience.
25/02/2025

Need someone to talk to?
Our support includes an active listening service and telephone helpline.