In the course of normal life there is a continuous recycling process of building new molecules and breaking down old ones. The breakdown and recycling process requires a series of special biochemical tools called enzymes. Beta-galactosidase breaks down several molecules, including a substance called GM1 ganglioside. People with GM1 gangliosidosis cannot make enough beta-galactosidase, without sufficient levels of beta-galactosidase the normal functioning of nerve cells in the brain is affected. Build-up of GM1 ganglioside can lead to harmful levels in many tissues and organs and results in progressive damage causing many of the signs and symptoms of GM1 gangliosidosis.
In general, the severity of GM1 gangliosidosis is related to the level of beta-galactosidase activity. People with higher enzyme activity levels usually have milder signs and symptoms than those with lower activity levels because they have less accumulation of GM1 ganglioside within the body.
The disease has traditionally been classified into three major types based on the age at which signs and symptoms first appear.
Type I, where signs and symptoms become apparent by 6 months and this is the most severe form.
Type II, where signs and symptoms become apparent from 18 months to around 5 years as is the intermediate form of the disease.
Type III, the mildest and chronic form seen during adulthood.
GM1 gangliosidosis represents a continuous spectrum of symptoms and significant overlapping features from type I to type III differing in severity.
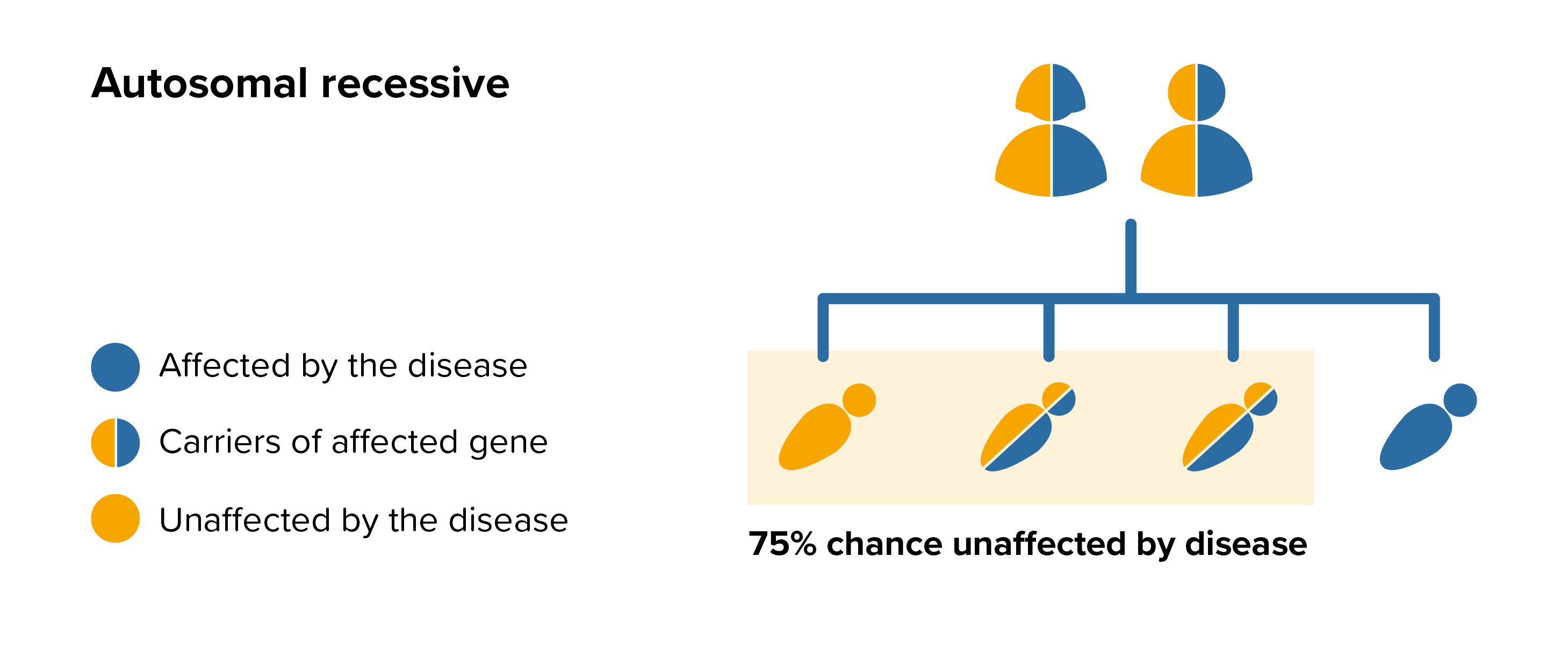 All parents of children with GM1 gangliosidosis can benefit from genetic counselling, the counsellor can provide advice on the risk to close relatives and to suggest whether the wider family should be informed. To find out during a pregnancy, if the baby is affected by GM1 gangliosidosis, screening tests can be arranged early on during a pregnancy for those families who already have a child with GM1 gangliosidosis. Where only one parent is a carrier, they can opt for carrier screening but it is not 100% reliable or accurate and is not possible in all cases.
All parents of children with GM1 gangliosidosis can benefit from genetic counselling, the counsellor can provide advice on the risk to close relatives and to suggest whether the wider family should be informed. To find out during a pregnancy, if the baby is affected by GM1 gangliosidosis, screening tests can be arranged early on during a pregnancy for those families who already have a child with GM1 gangliosidosis. Where only one parent is a carrier, they can opt for carrier screening but it is not 100% reliable or accurate and is not possible in all cases. 




