Research is ongoing to determine the cause.
Geleophysic dysplasia is a very rare progressive disease which comes under a group of diseases related to the mucopolysaccharidoses.
Read on for information about the condition or see the latest updates and resources.
Frequently asked questions
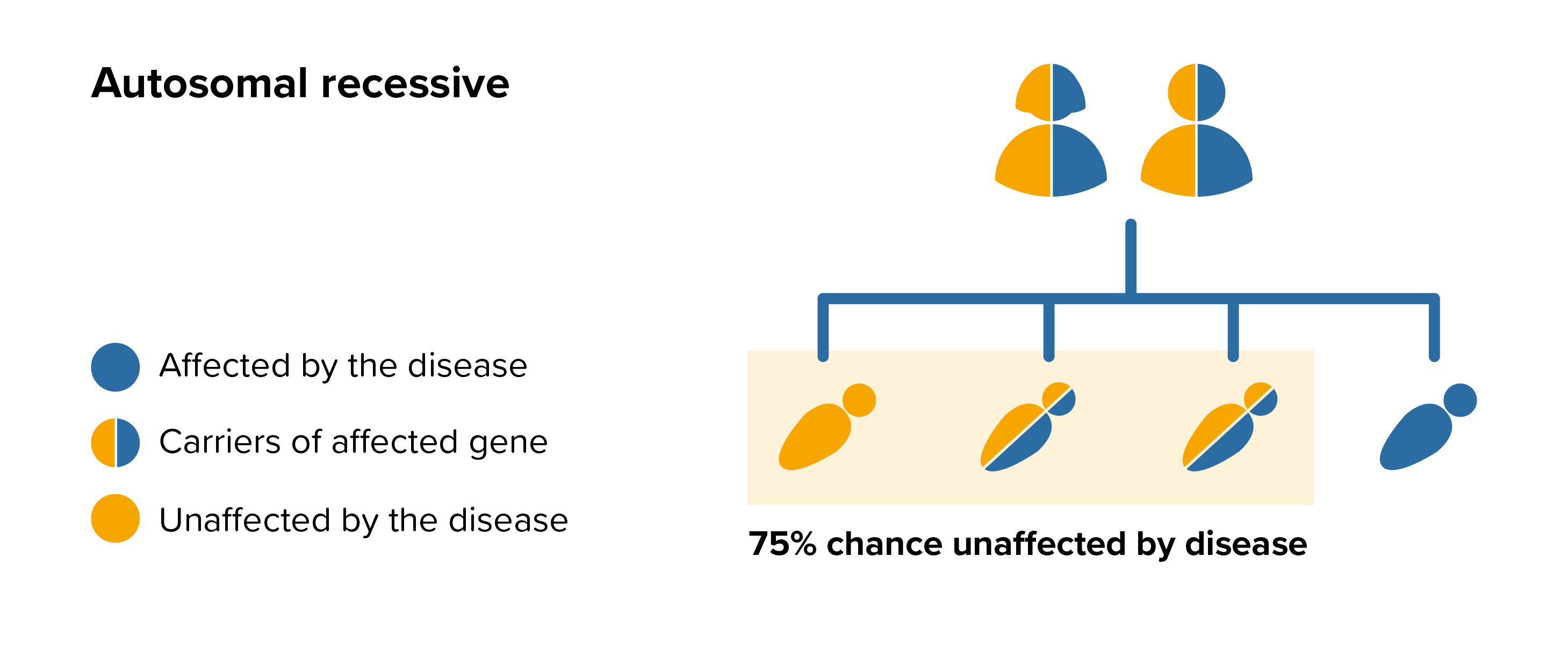
Geleophysic dysplasia is an autosomal recessive disease this means that both parents must carry the same affected gene and each pass this same affected gene to their child.
People probably carry from 5 to 10 genes with mutations in each of their cells. Problems happen when the particular gene is dominant or when a mutation is present in both copies of a recessive gene pair. Genes are the unique set of instructions inside our bodies that make each of us an individual. They are the blueprint for our growth and development, as well as controlling how our bodies function.
Genes are carried on structures called chromosomes and it is usual to have 23 pairs. A child will inherit half of the chromosomes from the mother and the other half from the father resulting in 23 pairs. 22 of these pairs look the same in both males and females. Pair 23 are the sex chromosomes, and this is the pair that differ between females and males. The X chromosome is inherited from the mother and the Y chromosome is inherited from the father. More information about inheritance is available in our publication.
For each pregnancy the chances of a baby inheriting geleophysic dysplasia are completely independent of whether a previous child was affected with geleophysic dysplasia. With each pregnancy there is a 1 in 4 chance that the baby will be affected by geleophysic dysplasia.
 All parents of children with geleophysic dysplasia can benefit from genetic counselling, the counsellor can provide advice on the risk to close relatives and to suggest whether the wider family should be informed. To find out during a pregnancy, if the baby is affected by geleophysic dysplasia, screening tests can be arranged early on during a pregnancy for those families who already have a child with geleophysic dysplasia.
All parents of children with geleophysic dysplasia can benefit from genetic counselling, the counsellor can provide advice on the risk to close relatives and to suggest whether the wider family should be informed. To find out during a pregnancy, if the baby is affected by geleophysic dysplasia, screening tests can be arranged early on during a pregnancy for those families who already have a child with geleophysic dysplasia.
Where only one parent is a carrier, they can opt for carrier screening but it is not 100% reliable or accurate and is not possible in all cases. Amniocentesis and chorionic villus sampling are both available during the pregnancy to find out if the baby is affected by geleophysic dysplasia.
It is estimated that nearly 6% of the UK population (around 3.5million people) will be affected by a rare disease at some point in their lives. A single rare disease may affect up to about 30,000 people however the vast majority of rare diseases affect far fewer than this.
Fewer than 30 cases of geleophysic dysplasia have been reported.
Children with geleophysic dysplasia have distinctive facial features. Often a shortened nose, full cheeks and abnormally large distance between the eyes. A common feature is a long flat philtrum, this is the vertical groove between the base of the nose and the border of the upper lip, the upper lip is thin. Overall children with geleophysic dysplasia are short in stature and have short hands and feet.
The skin is thickened as are the heart valves, which often lead to an early death. Breathing is complicated by a narrowed windpipe. The liver is enlarged which can further add strain to breathing by restricting the lugs to really expand for taking big breathes.
Contractions of leg muscles and Achilles tendon lead to tip toe walking with overall progressive joint limitation and shortening of the muscles.
The brain remains unaffected and intellectual ability is normal.
Major issues are likely to be present in the first year of life and it is likely that heart and lung involvement can result in death before the age 5.
At present there is treatment for symptoms as they arise, but no cure for the underlying disease. More information on supportive care treatments for people with MPS and related diseases can be found in the treatments section.
For an up-to-date list of current UK based trials taking place visit Be Part of Research (resource provided by the National Institute for Health Research). For an international search visit Clinical Trials (resource provided by the U.S. National Library of Medicine).
This resource provides information on trial status including recruiting, completed or withdrawn and worldwide trial locations. To find out more about past or current trials speak to your doctor and learn about the risks and potential benefits.
The MPS Society is the only UK charity at the forefront of supporting people and families affected by MPS and related diseases. Our extensive support services offer you a wide range of support and resources.
The team can advise and sign post you to adequate needs-led support and services in your local area as well as social care, home adaptions, education and much more.
The support team can visit you in your home and provide you with vital support.
Get involved and support us in the community, volunteer or support fundraising; we are a small charity but with your support we can continue to offer a highly valued and essential service.

Need someone to talk to?
Our support includes an active listening service and telephone helpline.
Latest updates

News
Christine Lavery Memorial Fund opens a call for applications for summer vacation studentships 2023
The MPS Society are delighted to announce that we are launching the Christine Lavery Memorial Fund on 21 November, Christine’s birthday,...
21/11/2022

Blog
Have your say to improve services for ultra-rare diseases
If you are affected by an ultra-rare disease we need your help to make support change and improve your care and services.
29/07/2020
Latest resources

Be prepared: emergency information for admission to hospital
Emergency information for admission to hospital that you should have to hand.

Covid
Please find below some updated information from our adult clinical centres. The clinical centres are working tirelessly to manage the...

Portacaths - To Port or Not to Port by Dr Fiona Stewart
MPS National Conference 2019 Portacaths - To Port or Not to Port by Dr Fiona Stewart Consultant in Medical Genetics, Clinical Genetics at...